„Business Insider Deutschland
Erdogan entlässt Notenbankchef und die türkische Lira stürzt ab
Christoph Damm vor 37 Min.
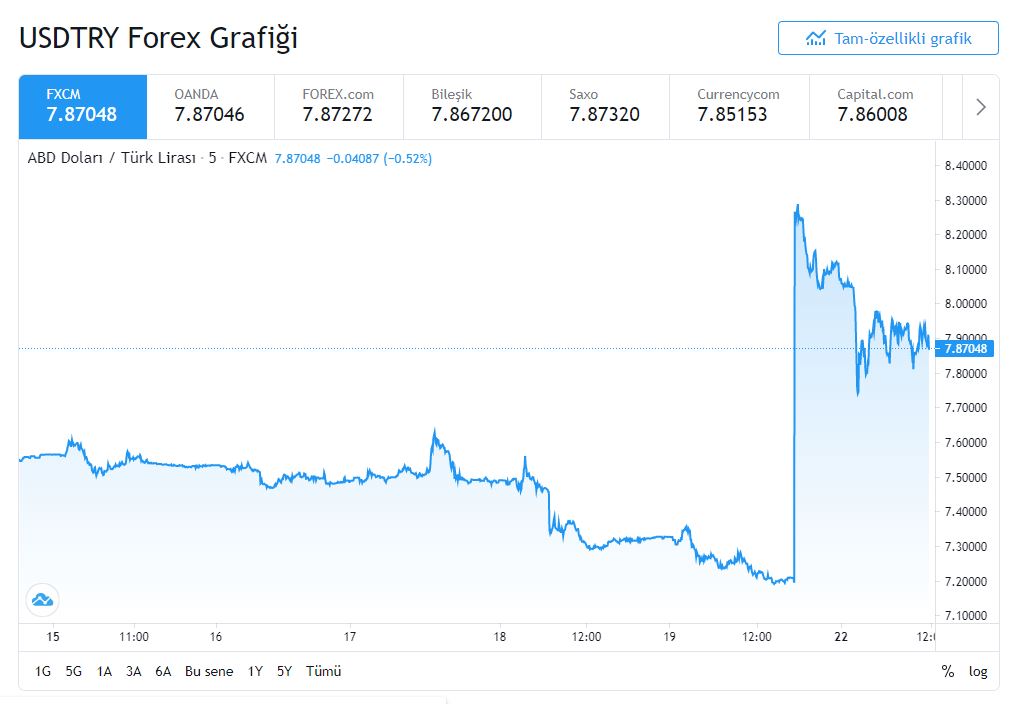
Was viele Beobachter befürchteten, ist nun eingetreten: Recep Tayyip Erdogan, Präsident der Türkei, hat Naci Agbal, Chef der türkischen Notenbank, entlassen. Infolge dessen stürzte die türkische Lira ab, am Montag fiel sie zum Dollar zeitweise um mehr als zehn Prozent 8,13 Lira ab.“
https://www.msn.com/de-de/finanzen/top-stories/erdogan-entl%c3%a4sst-notenbankchef-und-die-t%c3%bcrkische-lira-st%c3%bcrzt-ab/ar-BB1ePDOk?ocid=msedgntp
AMA ESAS ÖNEMLI OLAN BU…
Science ulan Science…
Nedir bilir misin?
„Studie sorgt für Aufsehen: Gibt es Corona schon viel länger, als wir alle dachten?
express vor 2 Std.
Das ist mal ein echter Hammer: Der Corona-Erreger Sars-CoV-2 hat einer Studie zufolge möglicherweise bereits Monate vor den ersten öffentlich bekannt gewordenen Krankheitsfällen existiert.“
https://www.msn.com/de-de/nachrichten/panorama/studie-sorgt-f%C3%BCr-aufsehen-gibt-es-corona-schon-viel-l%C3%A4nger-als-wir-alle-dachten/ar-BB1ePAr4?ocid=msedgntp
„REPORT
Timing the SARS-CoV-2 index case in Hubei province
View ORCID ProfileJonathan Pekar1,2, View ORCID ProfileMichael Worobey3,*, View ORCID ProfileNiema Moshiri4, View ORCID ProfileKonrad Scheffler5, View ORCID ProfileJoel O. Wertheim6,*
See all authors and affiliations
Science 18 Mar 2021:
eabf8003
DOI: 10.1126/science.abf8003
Article
Figures & Data
Info & Metrics
eLetters
PDF
Abstract
Understanding when SARS-CoV-2 emerged is critical to evaluating our current approach to monitoring novel zoonotic pathogens and understanding the failure of early containment and mitigation efforts for COVID-19. We employed a coalescent framework to combine retrospective molecular clock inference with forward epidemiological simulations to determine how long SARS-CoV-2 could have circulated prior to the time of the most recent common ancestor. Our results define the period between mid-October and mid-November 2019 as the plausible interval when the first case of SARS-CoV-2 emerged in Hubei province. By characterizing the likely dynamics of the virus before it was discovered, we show that over two-thirds of SARS-CoV-2-like zoonotic events would be self-limited, dying out without igniting a pandemic. Our findings highlight the shortcomings of zoonosis surveillance approaches for detecting highly contagious pathogens with moderate mortality rates.
SIGN UP FOR THE SCIENCE eTOC
Get the latest issue of Science delivered right to you!
Email
Email address*
In late-December of 2019, the first cases of COVID-19, the disease caused by SARS-CoV-2, were described in the city of Wuhan in Hubei province, China (1, 2). The virus quickly spread within China (3). The cordon sanitaire that was put in place in Wuhan on 23 January 2020 and mitigation efforts across China eventually brought about an end to sustained local transmission. In March and April 2020, restrictions across China were relaxed (4). By then, however, COVID-19 was a pandemic (5).
A concerted effort has been made to determine when the virus first began transmitting among humans by retrospectively diagnosing the earliest cases of COVID-19. Both epidemiological and phylogenetic approaches suggest an emergence of the pandemic in Hubei province at some point in late-2019 (2, 6, 7). The first described cluster of COVID-19 was associated with the Huanan Seafood Wholesale Market in late-December 2019, and the earliest sequenced SARS-CoV-2 genomes came from this cluster (8, 9). However, this market cluster is unlikely to have marked the beginning of the pandemic, as COVID-19 cases from early December lacked connections to the market (7). The earliest such case in the scientific literature is from an individual retrospectively diagnosed on 01 December 2019 (6). Notably, however, newspaper reports document retrospective COVID-19 diagnoses recorded by the Chinese government going back to 17 November 2019 in Hubei province (10). In fact, these reports detail daily retrospective COVID-19 diagnoses through the end of November, suggesting that SARS-CoV-2 was actively circulating for at least a month before it was discovered.
Molecular clock phylogenetic analyses have inferred the time of most recent common ancestor (tMRCA) of all sequenced SARS-CoV-2 genomes to be in late November or early December 2019, with uncertainty estimates typically ranging back to October 2019 (7, 11, 12). Crucially, though, this tMRCA is not necessarily equivalent to the date of zoonosis or index case infection (13, 14), because coalescent processes can prune basal viral lineages before they have the opportunity to be sampled, potentially pushing SARS-CoV-2 tMRCA estimates forward in time from the index case by days, weeks, or months. For a point of comparison, consider the zoonotic origins of the HIV-1 pandemic, whose tMRCA in the early 20th century coincides with the urbanization of Kinshasa, in the Congo (15, 16), but whose cross-species transmission from a chimpanzee reservoir occurred in southeast Cameroon, likely predating the tMRCA of sampled HIV-1 genomes by many years (17). Despite this important distinction, the tMRCA has been frequently conflated with the date of the index case infection in the SARS-CoV-2 literature (7, 18, 19).
Here, we combine retrospective molecular clock analysis in a coalescent framework with a forward compartmental epidemiological model to estimate the timing of the SARS-CoV-2 index case in Hubei province. The inferred dynamics during these unobserved early days of SARS-CoV-2 highlight challenges in detecting and preventing nascent pandemics.
We first explored the evolutionary dynamics of the first wave of SARS-CoV-2 infections in China. We used Bayesian phylodynamics (20) to reconstruct the underlying coalescent processes using a Bayesian Skyline approach for 583 SARS-CoV-2 complete genomes, sampled in China between when the virus was first discovered at the end of December 2019 and the last of the non-reintroduced circulating virus in April 2020. Applying a strict molecular clock, we inferred an evolutionary rate of 7.90×10−4 substitutions/site/year (95% highest posterior density [HPD]: 6.64×10−4–9.27×10−4). The tMRCA of these circulating strains was inferred to fall within a 34-day window with a mean of 09 December 2019 (95% HPD: 17 November–20 December) (Fig. 1). This estimate accounts for the many disparate inferred rooting orientations [see supplementary material and described previously (21)]. Notably, 78.7% of the posterior density post-dates the earliest published case on 01 December, and 95.1% post-dates the earliest reported case on 17 November. Relaxing the molecular clock provides a similar tMRCA estimate, as does applying a Skygrid coalescent approach (fig. S1). The recency of this tMRCA estimate in relation to the earliest documented COVID-19 cases obliges us to consider the possibility that this tMRCA does not capture the index case and that SARS-CoV-2 was circulating in Hubei province prior to the inferred tMRCA.
Download high-res image
Open in new tab
Download Powerpoint
Fig. 1 Posterior distribution for the time of the most recent common ancestor (tMRCA) of 583 sampled SARS-CoV-2 genomes circulating in China between December 2019 and April 2020.
Inference was performed using a strict molecular clock and a Bayesian Skyline coalescent prior. Shaded area denotes 95% HPD. Long-dashed line is 17 November 2019, and short-dashed line is 01 December 2019.
If the tMRCA post-dates the earliest documented cases, then the earliest diverged SARS-CoV-2 lineages must have gone extinct (Fig. 2). As these early basal lineages disappeared, the tMRCA of the remaining lineages would move forward in time (fig. S2). Thus, we interrogated the posterior trees sampled from the phylodynamic analysis to determine if this time of coalescence had stabilized prior to the sequencing of the first SARS-CoV-2 genomes on 24 December 2019 or if this process of basal lineage loss was ongoing in late-December/early-January. Importantly, these basal lineages need not be associated with specific mutations, as the phylodynamic inference reconstructs the coalescent history, not the mutational history (20).
Download high-res image
Open in new tab
Download Powerpoint
Fig. 2 Hypothetical coalescent scenarios depicting how the time between index case infection and time of stable coalescence can vary based on stochastic extinction events of basal viral lineages.
Coalescence can occur within or contemporaneously with the index case (upper left) or in cases infected later in the course of the epidemic with one (upper right) or more (lower left) basal lineages going extinct. In extreme cases, the epidemic can persist at low levels for a long time before stable coalescence (lower right).
We find only weak evidence for basal lineage loss between 24 December 2019 and 13 January 2020 (fig. S3A). The root tMRCA is within 1 day of the tMRCA of virus sampled on or after 01 January 2020 in 78.5% of posterior samples (fig. S3B). The tMRCA of genomes sampled on or after 01 January 2020 is 3 days younger than the tMRCA of all sampled genomes. In contrast, the mean tMRCA does not change when considering genomes sampled on or after 01 January 2020 versus on or after 13 January 2020. This consistency indicates a stabilization of coalescent processes at the start of 2020 when there had been an estimated 1000 total people infected with SARS-CoV-2 in Wuhan (22). Nonetheless, to account for the weak signal of a delay in reaching a stable coalescence (i.e., the point in time at which basal lineages cease to be lost), we identified the tMRCA for all viruses sampled on or after 01 January 2020 (i.e., at the time of stable coalescence) for each tree in the posterior sample.
Phylogenetic analysis alone cannot tell us how long SARS-CoV-2 could have circulated in Hubei province before the tMRCA. To answer this question, we performed forward epidemic simulations (23). These simulations were initiated by a single index case using a compartmental epidemiological model across scale-free contact networks (mean number of contacts = 16). This compartmental model was previously developed to describe SARS-CoV-2 transmission dynamics in Wuhan (22). This model, termed SAPHIRE, includes compartments for susceptible (S), exposed (E), presymptomatic (P), unascertained (A), ascertained (I), hospitalized (H), and removed (R) individuals. Our simulations used parameters from the time period prior to COVID-19 mitigation efforts, from 01 January through 22 January 2020 (table S1), based on Hao et al. (22). We analyzed 1000 epidemic simulations that resulted in ≥1000 total infected people. These simulated epidemics had a median doubling time of 4.1 days (95% range across simulations: 2.7-6.7), matching pre-mitigation incidence trends in Wuhan (table S2).
We simulated coalescent processes across the transmission network to determine the tMRCA of the virus at the end of the simulation. This approach allowed us to determine the distribution of the expected number of days between index case infection and the stable coalescence (i.e., tMRCA) (Fig. 2). The median number of days between index case infection and this tMRCA was 8.0 days (95% range: 0.0 and 41.5 days) (Fig. 3A). The median time between index case infection and the first person exiting the presymptomatic phase (i.e., ascertained or unascertained infection) was 5.7 days (95% range: 0.9 to 15.7 days).
Download high-res image
Open in new tab
Download Powerpoint
Fig. 3 Forward simulations estimating the timing of the index case in Hubei province.
(A) Days between index case infection and stable coalescence in forward compartmental epidemic simulations (n = 1000). (B) Days between index case infection and stable coalescence after rejection sampling and (C) posterior distribution for date of index case infection, conditioned on an ascertained case by 17 November 2019 denoted by long-dashed line. (D and E) Epidemic simulation, conditioned on an ascertained case by 01 December 2019 denoted by short-dashed line. Two-phase epidemic (F) days between index case carrying less fit variant and adaptation (n = 2000), (G) days between adaptation and stable coalescence (n = 1000), and (H) posterior distribution for date of index case infection, conditioned on an ascertained case by 17 November 2019. (I to K) Two-phase epidemic conditioned on an ascertained case by 01 December 2019. Gray dashed lines indicate median estimates.
As a robustness check, we also simulated epidemics with more densely and more sparsely connected contact networks (mean 26 and 10 contacts, respectively), rescaling the per-contact transmission rate to maintain empirical epidemic growth dynamics. We also explored the effects of faster (mean: 3.1 days; 95% range: 2.0 to 5.1 days) and slower (mean: 5.3 days; 95% range: 3.6 to 7.5 days) epidemic doubling times (table S2). Slower transmission rates led to more days between the index case and the stable coalescence, but modifying the density of the contact network had minimal effect on this time interval (fig. S4).
To estimate the date of infection for the index case in Hubei province, we combined the retrospective molecular clock analysis with the forward epidemic simulations (fig. S5). We identified the stable tMRCA in the posterior trees as an anchor to the real-world calendar dates and then extended this date back in time according to the number of days between the index case infection and the time of stable coalescence from the compartmental epidemic simulations. However, a random sample of tMRCAs and days from index case infection to coalescence will not produce epidemiologically meaningful results, because many of these combinations do not precede the earliest dates of reported COVID-19 cases. Therefore, we implemented a rejection sampling-based approach to generate a posterior distribution of dates of infection for the Hubei index case, conditioning on at least one individual who has progressed past the pre-symptomatic stage in the simulated epidemic before the date of the first reported COVID-19 case (see methods; fig. S6).
In our primary analysis, we assume that 17 November represents the first documented case of COVID-19 (ascertained or unascertained in the SAPHIRE model). Under this assumption, the median number of days between index case infection and stable coalescence after rejection sampling is 37 days (95% HPD: 12 to 55 days) (Fig. 3B). Consequently, the index case in Hubei likely contracted SARS-CoV-2 on or around 04 November 2019 (95% upper HPD on 15 October; 99% upper HPD on 07 October) (Fig. 3C).
This timeframe for the Hubei index case is robust (fig. S7). Epidemic simulations with faster or slower transmission rates and more or less densely connected contact networks produce similar date estimates. Further, using the root tMRCA from the sampled posterior trees, rather than adjusting for the shifting coalescence between 24 December 2019 and 01 January 2020, produces a median date of 03 November (95% upper HPD 14 October; 99% upper HPD 04 October). Incorporating a relaxed molecular clock or adjusting the coalescent prior assumption (Skyline vs. Skygrid) also had minimal effect.
If we enforce that there be at least one ascertained case in our simulations prior to 17 November 2019, the median date of the Hubei index case is pushed back about a week to 28 October (95% upper HPD 11 October; 99% upper HPD 05 October) (fig. S7). However, the distinction between ascertained and unascertained in the original SAPHIRE model was meant to reflect the probability of missed diagnoses in January 2020 of the Wuhan epidemic and does not account for the investigations that resulted in retrospective diagnoses in November and December 2019.
If we discount the reported evidence of retrospective COVID-19 diagnoses throughout the end of November and instead take 01 December as representing the first confirmed case of COVID-19, then the median time between index case infection and stable coalescence after rejection sampling is 23 days (95% range: 1 to 47 days) (Fig. 3D). Under this scenario, the index case in Hubei would have contracted SARS-CoV-2 on or around 17 November 2019 (95% upper HPD 24 October; 99% upper HPD 13 October) (Fig. 3E). Similar dates are inferred with a relaxed molecular clock and conditioning on an ascertained infection by 01 December (fig. S7).
It is reasonable to postulate that the variant of SARS-CoV-2 that first emerged was less fit than the variant that spread through China and that evolutionary adaptation was critical to its establishment in humans (12). Therefore, we simulated two-phase epidemics wherein the index case was infected with a less fit variant (i.e., half as transmissible) that went extinct but not before giving rise to a mutant strain matching the transmission dynamics estimated in Wuhan (figs. S8 and S9; supplementary material). If we condition on an ascertained or unascertained COVID-19 case (due to either the original or adapted variant) by 17 November, the original variant transmits for a median of 5 days (95% HPD: 0-36 days) before the adapted strain emerges (Fig. 3F); this adapted strain then circulates for a median of 28 days (95% HPD: 0-52 days) before reaching a stable coalescence (Fig. 3G). In this scenario, the index case in Hubei would have likely contracted an unobserved variant of SARS-CoV-2 on 05 November 2019 (95% upper HPD on 11 October; 99% upper HPD on 25 September) (Fig. 3H). If we again discount the reported evidence COVID-19 in November and take 01 December as the first confirmed case, then the virus would spend less time in humans across both phases of the early epidemic (Fig. 3, I and J) and the index case in Hubei would have acquired SARS-CoV-2 on 18 November 2019 (95% upper HPD on 20 October; 99% upper HPD on 05 October) (Fig. 3K). As in the primary analysis, the inferred date of the index case in the two-phase epidemic was robust to varying model assumptions (fig. S10), including the amount by which viral fitness differed between the two phases (fig. S18; supplementary material).
These one-phase and two-phase epidemic simulations both suggest that the tMRCA is not representative of the emergence of SARS-CoV-2. In the primary analysis, the index case remains infected at the tMRCA (as in Fig. 2, upper right panel) in less than 1.1% of simulated epidemics (table S3). In the two-phase epidemics, the index case is still infected at the tMRCA in 2.2% of simulations, likely due to an increased variance in the time between index case and tMRCA. The initial, less fit variant persisted until the tMRCA in 17% of simulated epidemics and until 01 January 2020 in 3.7% of simulated epidemics. However, when this less fit variant persisted, it was represented by only a single infected individual at the tMRCA; this low frequency suggests that even if this less fit variant did exist, it could have easily been missed in early genome sequencing efforts. In the two-phase epidemic, the first ascertained (or unascertained) case was due to the less fit variant in around two-thirds of simulated epidemics.
By anchoring our epidemic simulations to specific tMRCA estimates, we can reconstruct a plausible range for the number of SARS-CoV-2 infections prior to the discovery of the virus (Fig. 4A). The median number of individuals infected with SARS-CoV-2 in our primary analysis is below 1 until 04 November. On 17 November, the median number infected is 4 individuals (95% HPD: 1-13) and reaches 9 (95% HPD: 2-26) on 01 December. These values are generally robust to model specifications, molecular clock method, and date of first COVID-19 case (table S4 and fig. S11). The two-phase epidemics tend to exhibit similar growth patterns (Fig. 4B, fig. S12, and table S5). Notably, we do not see any evidence for an increase in hospitalizations until mid-to-late December, even when we increase the virulence of the less-fit variant in the two-phase epidemics or increase the probability of hospitalization prior to the stable coalescence (supplementary material; figs. S13 and S14).
Download high-res image
Open in new tab
Download Powerpoint
Fig. 4 Epidemic growth in compartmental simulations.
(A) Total estimated people infected in late-2019. Dark purple shading is central 50% HPD, intermediate purple shading is central 95% HPD, and light purple is central 99% HPD. (B) Total estimated people infected in late-2019 for a two-phase epidemic. (C) The number of people infected over time in a sample of epidemic simulations that established (purple; n = 30) and went extinct (gray; n = 70). The y-axis transitions to log-scale once ≥10 people are infected at any given time. The lower panel shows the proportion of simulations that still have ≥1 infected individual over time (persisting epidemics in purple; extinct epidemics in gray). (D) A sample (n = 10) of two-phase epidemic simulations transitioning from less-fit Phase 1 (blue) to more-fit Phase 2 (purple). Each line represents a single simulation and its transition over time. The lower panel shows the average proportion of Phase 1 to Phase 2 infected individuals over time.
Empirical observation throughout the SARS-CoV-2 pandemic has shown the outsized role of superspreading events in the propagation of SARS-CoV-2 (24–27), wherein the average infected person does not transmit the virus. Our results suggest the same dynamics likely influenced the initial establishment of SARS-CoV-2 in humans, as only 29.7% of simulated epidemics from the primary analysis went on to establish self-sustaining epidemics. The remaining 70.3% of epidemics went extinct (Fig. 4C). Epidemics that went extinct typically produced only 1 infection (95% range: 1-9) and never more than 44 infections total or 14 infections at any given time (table S2). The median failed epidemic went extinct by day 8. As the contact network became more or less densely connected, the number of epidemics that went extinct was similar: 68.3% and 69.4%, respectively (table S2). However, the percentage of extinct epidemics increased as the transmission rate decreased (80.5%) and decreased as the transmission rate increased (53.6%). In the two-phase epidemic, this original less fit variant went extinct by day 9 (95% HPD: 2-52) and produced a median of 1 infection (95% HPD: 1-13) (Fig. 4D and table S6).
The overdispersed nature of SARS-CoV-2 transmission patterns favors its persistence, as epidemics simulated over random contact networks (27) that are not characterized by superspreading events tended to go extinct more frequently: 83.7% of the time. Furthermore, the large and highly connected contact networks characterizing urban areas seem critical to the establishment of SARS-CoV-2. When we simulated epidemics where the number of connections was reduced by 50% or 75% (without rescaling per-contact transmissibility), to reflect emergence in a rural community, the epidemics went extinct 94.5% or 99.6% of the time, respectively.
Our results highlight the unpredictable dynamics that characterized the earliest days of the COVID-19 pandemic. The successful establishment of SARS-CoV-2 post-zoonosis was far from certain, as more than two-thirds of simulated epidemics quickly went extinct. It is highly probable that SARS-CoV-2 was circulating in Hubei province at low levels in early-November 2019 and possibly as early as October 2019, but not earlier. Nonetheless, the inferred prevalence of this virus was too low to permit its discovery and characterization for weeks or months. By the time COVID-19 was first identified, the virus had firmly established itself in Wuhan. This delay highlights the difficulty in surveillance for novel zoonotic pathogens with high transmissibility and moderate mortality rates.
The high extinction rates we inferred suggest that spillover of SARS-CoV-2-like viruses may be frequent, even if pandemics are rare (28). Further, the same dynamics that characterized the establishment of SARS-CoV-2 in Hubei province may have played out all over the world as the virus was repeatedly introduced, but only occasionally took hold (29, 30). The reports of cases in December 2019 and January 2020 in France and California that did not establish sustained transmission fit this pattern (31–33). However, our results suggest that PCR evidence of SARS-CoV-2 in wastewater outside of China before November 2019 is unlikely to be valid (34) and the suggestion of international spread in mid-November or early-December 2019 should be viewed with skepticism (35–37), given that our results suggests fewer than 20 people infected with SARS-CoV-2 at this time (table S4 and fig. S11). Our results also refute claims (38) of large numbers of patients requiring hospitalization due to COVID-19 in Hubei province prior to December 2019 (figs. S13 and S14). Nevertheless, SARS-CoV-2 may be detectable should archived wastewater samples or other biomaterials from Hubei province exist from early-to-mid November 2019, and incorporating these types of data in our model could further refine our timing estimates. Moreover, wastewater detection may present the best chance of early detection of future pandemics during the early phase of spread where we estimate very low numbers of infections (39).
Even though all of the earliest documented cases of COVID-19 were found in Hubei province, we cannot discount the possibility that the index case initially acquired the virus elsewhere. Nonetheless, our dating inference is insensitive to geography. Further, our results suggest that if the virus first emerged in a rural community, it would have needed to migrate to an urban setting to avoid extinction. The lack of reports of COVID-19 elsewhere in China in November and early-December suggest Hubei province is the location where human-to-human transmission chains were first established.
The circumstances surrounding the emergence of SARS-CoV-2 in Hubei province remain shrouded. Although SARS-CoV-2 is repeatedly adapting to spread among humans (40, 41), our findings do not reveal whether the virus that first emerged was less fit than the virus that spread throughout China. Nevertheless, the inferred timing of the index case is generally similar in both of these scenarios, because less-fit viruses in our simulations that went extinct tended to do so very quickly. Importantly, it is yet unknown whether the virus emerged directly from its animal reservoir, presumably horseshoe bats (42, 43), or first circulated in and possibly adapted to an intermediate host. Our estimates for the timing of the Hubei index case further distance this individual from the outbreak at the Huanan Seafood Wholesale Market. Finding the animal reservoir, or hypothetical intermediate host, will help to further narrow down the date, location, and circumstances of the original SARS-CoV-2 infection in humans. However, even in the absence of that information, coalescent-based approaches permit us to look back beyond the tMRCA and toward the earliest days of the COVID-19 pandemic. Although there was a pre-tMRCA fuse to the COVID-19 pandemic, it was almost certainly very short. This brief period of time suggests that future pandemics with similar characteristics to the COVID-19 pandemic permit only a narrow window for preemptive intervention.
Supplementary Materials
science.sciencemag.org/cgi/content/full/science.abf8003/DC1
Materials and Methods
Supplementary Text
Figs. S1 to S18
Tables S1 to S7
References (45–54)
MDAR Reproducibility Checklist
Data S1
https://creativecommons.org/licenses/by/4.0/
This is an open-access article distributed under the terms of the Creative Commons Attribution license, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
References and Notes
↵ N. Zhu, D. Zhang, W. Wang, X. Li, B. Yang, J. Song, X. Zhao, B. Huang, W. Shi, R. Lu, P. Niu, F. Zhan, X. Ma, D. Wang, W. Xu, G. Wu, G. F. Gao, W. Tan; China Novel Coronavirus Investigating and Research Team, A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020). doi:10.1056/NEJMoa2001017pmid:31978945CrossRefPubMedGoogle Scholar
↵ Q. Li, X. Guan, P. Wu, X. Wang, L. Zhou, Y. Tong, R. Ren, K. S. M. Leung, E. H. Y. Lau, J. Y. Wong, X. Xing, N. Xiang, Y. Wu, C. Li, Q. Chen, D. Li, T. Liu, J. Zhao, M. Liu, W. Tu, C. Chen, L. Jin, R. Yang, Q. Wang, S. Zhou, R. Wang, H. Liu, Y. Luo, Y. Liu, G. Shao, H. Li, Z. Tao, Y. Yang, Z. Deng, B. Liu, Z. Ma, Y. Zhang, G. Shi, T. T. Y. Lam, J. T. Wu, G. F. Gao, B. J. Cowling, B. Yang, G. M. Leung, Z. Feng, Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N. Engl. J. Med. 382, 1199–1207 (2020). doi:10.1056/NEJMoa2001316pmid:31995857CrossRefPubMedGoogle Scholar
↵World Health Organization, “Report of the WHO-China Joint Mission on Coronavirus Disease 2019 (COVID-19)”; www.who.int/publications/i/item/report-of-the-who-china-joint-mission-on-coronavirus-disease-2019-(covid-19).
↵ H. Fei, X. Yinyin, C. Hui, W. Ni, D. Xin, C. Wei, L. Tao, H. Shitong, S. Miaomiao, C. Mingting, S. Keshavjee, Z. Yanlin, D. P. Chin, L. Jianjun, The impact of the COVID-19 epidemic on tuberculosis control in China. Lancet Reg. Health West. Pac. 3, 100032 (2020). doi:10.1016/j.lanwpc.2020.100032CrossRefGoogle Scholar
↵World Health Organization, “WHO Director-General’s opening remarks at the media briefing on COVID-19 – 11 March 2020”; www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19—11-march-2020.
↵ C. Huang, Y. Wang, X. Li, L. Ren, J. Zhao, Y. Hu, L. Zhang, G. Fan, J. Xu, X. Gu, Z. Cheng, T. Yu, J. Xia, Y. Wei, W. Wu, X. Xie, W. Yin, H. Li, M. Liu, Y. Xiao, H. Gao, L. Guo, J. Xie, G. Wang, R. Jiang, Z. Gao, Q. Jin, J. Wang, B. Cao, Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020). doi:10.1016/S0140-6736(20)30183-5pmid:31986264CrossRefPubMedGoogle Scholar
↵ X. Zhang, Y. Tan, Y. Ling, G. Lu, F. Liu, Z. Yi, X. Jia, M. Wu, B. Shi, S. Xu, J. Chen, W. Wang, B. Chen, L. Jiang, S. Yu, J. Lu, J. Wang, M. Xu, Z. Yuan, Q. Zhang, X. Zhang, G. Zhao, S. Wang, S. Chen, H. Lu, Viral and host factors related to the clinical outcome of COVID-19. Nature 583, 437–440 (2020). doi:10.1038/s41586-020-2355-0pmid:32434211CrossRefPubMedGoogle Scholar
↵ F. Wu, S. Zhao, B. Yu, Y.-M. Chen, W. Wang, Z.-G. Song, Y. Hu, Z.-W. Tao, J.-H. Tian, Y.-Y. Pei, M.-L. Yuan, Y.-L. Zhang, F.-H. Dai, Y. Liu, Q.-M. Wang, J.-J. Zheng, L. Xu, E. C. Holmes, Y.-Z. Zhang, A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020). doi:10.1038/s41586-020-2008-3pmid:32015508CrossRefPubMedGoogle Scholar
↵ R. Lu, X. Zhao, J. Li, P. Niu, B. Yang, H. Wu, W. Wang, H. Song, B. Huang, N. Zhu, Y. Bi, X. Ma, F. Zhan, L. Wang, T. Hu, H. Zhou, Z. Hu, W. Zhou, L. Zhao, J. Chen, Y. Meng, J. Wang, Y. Lin, J. Yuan, Z. Xie, J. Ma, W. J. Liu, D. Wang, W. Xu, E. C. Holmes, G. F. Gao, G. Wu, W. Chen, W. Shi, W. Tan, Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 395, 565–574 (2020). doi:10.1016/S0140-6736(20)30251-8pmid:32007145CrossRefPubMedGoogle Scholar
↵J. Ma, “Coronavirus: China’s first confirmed Covid-19 case traced back to November 17.” South China Morning Post (2020); www.scmp.com/news/china/society/article/3074991/coronavirus-chinas-first-confirmed-covid-19-case-traced-back.
↵A. Rambaut, “Phylodynamic Analysis | 176 genomes | 6 Mar 2020.” Virological (2020); https://virological.org/t/phylodynamic-analysis-176-genomes-6-mar-2020/356.
↵ S. Duchene, L. Featherstone, M. Haritopoulou-Sinanidou, A. Rambaut, P. Lemey, G. Baele, Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 6, veaa061 (2020). doi:10.1093/ve/veaa061pmid:33235813CrossRefPubMedGoogle Scholar
↵ K. G. Andersen, A. Rambaut, W. I. Lipkin, E. C. Holmes, R. F. Garry, The proximal origin of SARS-CoV-2. Nat. Med. 26, 450–452 (2020). doi:10.1038/s41591-020-0820-9pmid:32284615CrossRefPubMedGoogle Scholar
↵L. du Plessis, O. Pybus, “Further musings on the tMRCA.” Virological (2020); https://virological.org/t/further-musings-on-the-tmrca/340.
↵ N. R. Faria, A. Rambaut, M. A. Suchard, G. Baele, T. Bedford, M. J. Ward, A. J. Tatem, J. D. Sousa, N. Arinaminpathy, J. Pépin, D. Posada, M. Peeters, O. G. Pybus, P. Lemey, HIV epidemiology. The early spread and epidemic ignition of HIV-1 in human populations. Science 346, 56–61 (2014). doi:10.1126/science.1256739pmid:25278604Abstract/FREE Full TextGoogle Scholar
↵ M. Worobey, M. Gemmel, D. E. Teuwen, T. Haselkorn, K. Kunstman, M. Bunce, J.-J. Muyembe, J.-M. M. Kabongo, R. M. Kalengayi, E. Van Marck, M. T. P. Gilbert, S. M. Wolinsky, Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature 455, 661–664 (2008). doi:10.1038/nature07390pmid:18833279CrossRefPubMedWeb of ScienceGoogle Scholar
↵ B. F. Keele, F. Van Heuverswyn, Y. Li, E. Bailes, J. Takehisa, M. L. Santiago, F. Bibollet-Ruche, Y. Chen, L. V. Wain, F. Liegeois, S. Loul, E. M. Ngole, Y. Bienvenue, E. Delaporte, J. F. Y. Brookfield, P. M. Sharp, G. M. Shaw, M. Peeters, B. H. Hahn, Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science 313, 523–526 (2006). doi:10.1126/science.1126531pmid:16728595Abstract/FREE Full TextGoogle Scholar
↵ T. Bedford, A. L. Greninger, P. Roychoudhury, L. M. Starita, M. Famulare, M.-L. Huang, A. Nalla, G. Pepper, A. Reinhardt, H. Xie, L. Shrestha, T. N. Nguyen, A. Adler, E. Brandstetter, S. Cho, D. Giroux, P. D. Han, K. Fay, C. D. Frazar, M. Ilcisin, K. Lacombe, J. Lee, A. Kiavand, M. Richardson, T. R. Sibley, M. Truong, C. R. Wolf, D. A. Nickerson, M. J. Rieder, J. A. Englund, J. Hadfield, E. B. Hodcroft, J. Huddleston, L. H. Moncla, N. F. Müller, R. A. Neher, X. Deng, W. Gu, S. Federman, C. Chiu, J. S. Duchin, R. Gautom, G. Melly, B. Hiatt, P. Dykema, S. Lindquist, K. Queen, Y. Tao, A. Uehara, S. Tong, D. MacCannell, G. L. Armstrong, G. S. Baird, H. Y. Chu, J. Shendure, K. R. Jerome; Seattle Flu Study Investigators, Cryptic transmission of SARS-CoV-2 in Washington state. Science 370, 571–575 (2020). doi:10.1126/science.abc0523pmid:32913002Abstract/FREE Full TextGoogle Scholar
↵ Q. Bi, Y. Wu, S. Mei, C. Ye, X. Zou, Z. Zhang, X. Liu, L. Wei, S. A. Truelove, T. Zhang, W. Gao, C. Cheng, X. Tang, X. Wu, Y. Wu, B. Sun, S. Huang, Y. Sun, J. Zhang, T. Ma, J. Lessler, T. Feng, Epidemiology and transmission of COVID-19 in 391 cases and 1286 of their close contacts in Shenzhen, China: A retrospective cohort study. Lancet Infect. Dis. 20, 911–919 (2020). doi:10.1016/S1473-3099(20)30287-5pmid:32353347CrossRefPubMedGoogle Scholar
↵ M. A. Suchard, P. Lemey, G. Baele, D. L. Ayres, A. J. Drummond, A. Rambaut, Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016 (2018). doi:10.1093/ve/vey016pmid:29942656CrossRefPubMedGoogle Scholar
↵ L. Pipes, H. Wang, J. P. Huelsenbeck, R. Nielsen, Assessing uncertainty in the rooting of the SARS-CoV-2 phylogeny. Mol. Biol. Evol. 10.1093/molbev/msaa316 (2020). doi:10.1093/molbev/msaa316pmid:33295605CrossRefPubMedGoogle Scholar
↵ X. Hao, S. Cheng, D. Wu, T. Wu, X. Lin, C. Wang, Reconstruction of the full transmission dynamics of COVID-19 in Wuhan. Nature 584, 420–424 (2020). doi:10.1038/s41586-020-2554-8pmid:32674112CrossRefPubMedGoogle Scholar
↵ N. Moshiri, M. Ragonnet-Cronin, J. O. Wertheim, S. Mirarab, FAVITES: Simultaneous simulation of transmission networks, phylogenetic trees and sequences. Bioinformatics 35, 1852–1861 (2019). doi:10.1093/bioinformatics/bty921pmid:30395173CrossRefPubMedGoogle Scholar
↵ R. Laxminarayan, B. Wahl, S. R. Dudala, K. Gopal, C. Mohan B, S. Neelima, K. S. Jawahar Reddy, J. Radhakrishnan, J. A. Lewnard, Epidemiology and transmission dynamics of COVID-19 in two Indian states. Science 370, 691–697 (2020). doi:10.1126/science.abd7672pmid:33154136Abstract/FREE Full TextGoogle Scholar
A. Endo, S. Abbott, A. J. Kucharski, S. Funk; Centre for the Mathematical Modelling of Infectious Diseases COVID-19 Working Group, Estimating the overdispersion in COVID-19 transmission using outbreak sizes outside China. Wellcome Open Res. 5, 67 (2020). doi:10.12688/wellcomeopenres.15842.3pmid:32685698CrossRefPubMedGoogle Scholar
X.-K. Xu, X. F. Liu, Y. Wu, S. T. Ali, Z. Du, P. Bosetti, E. H. Y. Lau, B. J. Cowling, L. Wang, Reconstruction of transmission pairs for novel coronavirus disease 2019 (COVID-19) in mainland China: Estimation of superspreading events, serial interval, and hazard of infection. Clin. Infect. Dis. 71, 3163–3167 (2020). doi:10.1093/cid/ciaa790pmid:32556265CrossRefPubMedGoogle Scholar
↵ D. C. Adam, P. Wu, J. Y. Wong, E. H. Y. Lau, T. K. Tsang, S. Cauchemez, G. M. Leung, B. J. Cowling, Clustering and superspreading potential of SARS-CoV-2 infections in Hong Kong. Nat. Med. 26, 1714–1719 (2020). doi:10.1038/s41591-020-1092-0pmid:32943787CrossRefPubMedGoogle Scholar
↵ H. Li, E. Mendelsohn, C. Zong, W. Zhang, E. Hagan, N. Wang, S. Li, H. Yan, H. Huang, G. Zhu, N. Ross, A. Chmura, P. Terry, M. Fielder, M. Miller, Z. Shi, P. Daszak, Human-animal interactions and bat coronavirus spillover potential among rural residents in Southern China. Biosaf. Health 1, 84–90 (2019). doi:10.1016/j.bsheal.2019.10.004pmid:32501444CrossRefPubMedGoogle Scholar
↵ M. Worobey, J. Pekar, B. B. Larsen, M. I. Nelson, V. Hill, J. B. Joy, A. Rambaut, M. A. Suchard, J. O. Wertheim, P. Lemey, The emergence of SARS-CoV-2 in Europe and North America. Science 370, 564–570 (2020). doi:10.1126/science.abc8169pmid:32912998Abstract/FREE Full TextGoogle Scholar
↵ L. du Plessis, J. T. McCrone, A. E. Zarebski, V. Hill, C. Ruis, B. Gutierrez, J. Raghwani, J. Ashworth, R. Colquhoun, T. R. Connor, N. R. Faria, B. Jackson, N. J. Loman, Á. O’Toole, S. M. Nicholls, K. V. Parag, E. Scher, T. I. Vasylyeva, E. M. Volz, A. Watts, I. I. Bogoch, K. Khan, D. M. Aanensen, M. U. G. Kraemer, A. Rambaut, O. G. Pybus; COVID-19 Genomics UK (COG-UK) Consortium, Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science 371, 708–712 (2021). doi:10.1126/science.abf2946pmid:33419936Abstract/FREE Full TextGoogle Scholar
↵ A. Deslandes, V. Berti, Y. Tandjaoui-Lambotte, C. Alloui, E. Carbonnelle, J. R. Zahar, S. Brichler, Y. Cohen, SARS-CoV-2 was already spreading in France in late December 2019. Int. J. Antimicrob. Agents 55, 106006 (2020). doi:10.1016/j.ijantimicag.2020.106006pmid:32371096CrossRefPubMedGoogle Scholar
X. Deng, W. Gu, S. Federman, L. du Plessis, O. G. Pybus, N. R. Faria, C. Wang, G. Yu, B. Bushnell, C.-Y. Pan, H. Guevara, A. Sotomayor-Gonzalez, K. Zorn, A. Gopez, V. Servellita, E. Hsu, S. Miller, T. Bedford, A. L. Greninger, P. Roychoudhury, L. M. Starita, M. Famulare, H. Y. Chu, J. Shendure, K. R. Jerome, C. Anderson, K. Gangavarapu, M. Zeller, E. Spencer, K. G. Andersen, D. MacCannell, C. R. Paden, Y. Li, J. Zhang, S. Tong, G. Armstrong, S. Morrow, M. Willis, B. T. Matyas, S. Mase, O. Kasirye, M. Park, G. Masinde, C. Chan, A. T. Yu, S. J. Chai, E. Villarino, B. Bonin, D. A. Wadford, C. Y. Chiu, Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science 369, 582–587 (2020). doi:10.1126/science.abb9263pmid:32513865Abstract/FREE Full TextGoogle Scholar
↵ M. A. Jorden, S. L. Rudman, E. Villarino, S. Hoferka, M. T. Patel, K. Bemis, C. R. Simmons, M. Jespersen, J. Iberg Johnson, E. Mytty, K. D. Arends, J. J. Henderson, R. W. Mathes, C. X. Weng, J. Duchin, J. Lenahan, N. Close, T. Bedford, M. Boeckh, H. Y. Chu, J. A. Englund, M. Famulare, D. A. Nickerson, M. J. Rieder, J. Shendure, L. M. Starita; CDC COVID-19 Response Team, Evidence for limited early spread of COVID-19 within the United States, January–February 2020. Morb. Mortal. Wkly. Rep. 69, 680–684 (2020). doi:10.15585/mmwr.mm6922e1pmid:32497028CrossRefPubMedGoogle Scholar
↵G. Chavarria-Miró, E. Anfruns-Estrada, S. Guix, M. Paraira, B. Galofré, G. Sánchez, R. M. Pintó, A. Bosch, Sentinel surveillance of SARS-CoV-2 in wastewater anticipates the occurrence of COVID-19 cases. medRxiv (2020); https://medrxiv.org/lookup/doi/10.1101/2020.06.13.20129627.
↵G. Fongaro, P. H. Stoco, D. S. Marques Souza, E. C. Grisard, M. E. Magri, P. Rogovski, M. A. Schörner, F. Hartmann Barazzetti, A. P. Christoff, L. F. V. de Oliveira, M. L. Bazzo, G. Wagner, M. Hernández, D. Rodriguez-Lázaro, SARS-CoV-2 in human sewage in Santa Catalina, Brazil, November 2019. medRxiv (2020); www.medrxiv.org/content/10.1101/2020.06.26.20140731v1.
G. La Rosa, P. Mancini, G. Bonanno Ferraro, C. Veneri, M. Iaconelli, L. Bonadonna, L. Lucentini, E. Suffredini, SARS-CoV-2 has been circulating in northern Italy since December 2019: Evidence from environmental monitoring. Sci. Total Environ. 750, 141711 (2021). doi:10.1016/j.scitotenv.2020.141711pmid:32835962CrossRefPubMedGoogle Scholar
↵ A. Amendola, S. Bianchi, M. Gori, D. Colzani, M. Canuti, E. Borghi, M. C. Raviglione, G. V. Zuccotti, E. Tanzi, Evidence of SARS-CoV-2 RNA in an oropharyngeal swab specimen, Milan, Italy, early December 2019. Emerg. Infect. Dis. 27, 648–650 (2021). doi:10.3201/eid2702.204632pmid:33292923CrossRefPubMedGoogle Scholar
↵E. O. Nsoesie, B. Rader, Y. L. Barnoon, L. Goodwin, J. Brownstein, Analysis of hospital traffic and search engine data in Wuhan China indicates early disease activity in the Fall of 2019. (2020); available at https://dash.harvard.edu/handle/1/42669767.
↵ J. Peccia, A. Zulli, D. E. Brackney, N. D. Grubaugh, E. H. Kaplan, A. Casanovas-Massana, A. I. Ko, A. A. Malik, D. Wang, M. Wang, J. L. Warren, D. M. Weinberger, W. Arnold, S. B. Omer, Measurement of SARS-CoV-2 RNA in wastewater tracks community infection dynamics. Nat. Biotechnol. 38, 1164–1167 (2020). doi:10.1038/s41587-020-0684-zpmid:32948856CrossRefPubMedGoogle Scholar
↵E. Volz, S. Mishra, M. Chand, J. C. Barrett, R. Johnson, L. Geidelberg, W. R. Hinsley, D. J. Laydon, G. Dabrera, Á. O’Toole, R. Amato, M. Ragonnet-Cronin, I. Harrison, B. Jackson, C. V. Ariani, O. Boyd, N. Loman, J. T. McCrone, S. Gonçalves, D. Jorgensen, R. Myers, V. Hill, D. K. Jackson, K. Gaythorpe, N. Groves, J. Sillitoe, D. P. Kwiatkowski, The COVID-19 Genomics UK (COG-UK) consortium, S. Flaxman, O. Ratman, S. Bhatt, S. Hopkins, A. Gandy, A. Rambaut, N. M. Ferguson, Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv (2021), .doi:10.1101/2020.12.30.20249034Abstract/FREE Full TextGoogle Scholar
↵ B. Korber, W. M. Fischer, S. Gnanakaran, H. Yoon, J. Theiler, W. Abfalterer, N. Hengartner, E. E. Giorgi, T. Bhattacharya, B. Foley, K. M. Hastie, M. D. Parker, D. G. Partridge, C. M. Evans, T. M. Freeman, T. I. de Silva, C. McDanal, L. G. Perez, H. Tang, A. Moon-Walker, S. P. Whelan, C. C. LaBranche, E. O. Saphire, D. C. Montefiori; Sheffield COVID-19 Genomics Group, Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 182, 812–827.e19 (2020). doi:10.1016/j.cell.2020.06.043pmid:32697968CrossRefPubMedGoogle Scholar
↵S. Lytras, J. Hughes, W. Xia, X. Jiang, D. L. Robertson, Exploring the natural origins of SARS-CoV-2. bioRxiv (2021); www.biorxiv.org/content/10.1101/2021.01.22.427830v2.
↵ M. F. Boni, P. Lemey, X. Jiang, T. T.-Y. Lam, B. W. Perry, T. A. Castoe, A. Rambaut, D. L. Robertson, Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 5, 1408–1417 (2020). doi:10.1038/s41564-020-0771-4pmid:32724171CrossRefPubMedGoogle Scholar
↵J. Pekar, J. Wertheim, Data for “Timing the SARS-CoV-2 Index Case in Hubei Province.” Data Dryad (2021); .doi:10.5061/dryad.4f4qrfjbmGoogle Scholar
↵ Y. Shu, J. McCauley, GISAID: Global initiative on sharing all influenza data – from vision to reality. Euro Surveill. 22, 30494 (2017). doi:10.2807/1560-7917.ES.2017.22.13.30494pmid:28382917CrossRefPubMedGoogle Scholar
B. Q. Minh, H. A. Schmidt, O. Chernomor, D. Schrempf, M. D. Woodhams, A. von Haeseler, R. Lanfear, IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020). doi:10.1093/molbev/msaa015pmid:32011700CrossRefPubMedGoogle Scholar
A. Rambaut, T. T. Lam, L. Max Carvalho, O. G. Pybus, Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2, vew007 (2016). doi:10.1093/ve/vew007pmid:27774300CrossRefPubMedGoogle Scholar
A. Rambaut, A. J. Drummond, D. Xie, G. Baele, M. A. Suchard, Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904 (2018). doi:10.1093/sysbio/syy032pmid:29718447CrossRefPubMedGoogle Scholar
M. S. Gill, P. Lemey, N. R. Faria, A. Rambaut, B. Shapiro, M. A. Suchard, Improving Bayesian population dynamics inference: A coalescent-based model for multiple loci. Mol. Biol. Evol. 30, 713–724 (2013). doi:10.1093/molbev/mss265pmid:23180580CrossRefPubMedWeb of ScienceGoogle Scholar
A.-L. Barabási, R. Albert, Emergence of scaling in random networks. Science 286, 509–512 (1999). doi:10.1126/science.286.5439.509pmid:10521342Abstract/FREE Full TextGoogle Scholar
J. Mossong, N. Hens, M. Jit, P. Beutels, K. Auranen, R. Mikolajczyk, M. Massari, S. Salmaso, G. S. Tomba, J. Wallinga, J. Heijne, M. Sadkowska-Todys, M. Rosinska, W. J. Edmunds, Social contacts and mixing patterns relevant to the spread of infectious diseases. PLOS Med. 5, e74 (2008). doi:10.1371/journal.pmed.0050074pmid:18366252CrossRefPubMedGoogle Scholar
F. D. Sahneh, A. Vajdi, H. Shakeri, F. Fan, C. Scoglio, GEMFsim: A stochastic simulator for the generalized epidemic modeling framework. J. Comput. Sci. 22, 36–44 (2017). doi:10.1016/j.jocs.2017.08.014CrossRefGoogle Scholar
O. Ratmann, E. B. Hodcroft, M. Pickles, A. Cori, M. Hall, S. Lycett, C. Colijn, B. Dearlove, X. Didelot, S. Frost, A. S. M. M. Hossain, J. B. Joy, M. Kendall, D. Kühnert, G. E. Leventhal, R. Liang, G. Plazzotta, A. F. Y. Poon, D. A. Rasmussen, T. Stadler, E. Volz, C. Weis, A. J. Leigh Brown, C. Fraser; PANGEA-HIV Consortium, Phylogenetic tools for generalized HIV-1 epidemics: Findings from the PANGEA-HIV Methods Comparison. Mol. Biol. Evol. 34, 185–203 (2017). doi:10.1093/molbev/msw217pmid:28053012CrossRefPubMedGoogle Scholar
↵ N. Moshiri, TreeSwift: A massively scalable Python tree package. SoftwareX 11, 100436 (2020). doi:10.1016/j.softx.2020.100436CrossRefGoogle Scholar
Acknowledgments: We gratefully acknowledge the authors from the originating laboratories and the submitting laboratories, who generated and shared via GISAID the viral genomic sequence data on which this research is based. A complete list acknowledging the authors who submitted data analyzed in this study can be found in data S1. We thank Jennifer Havens, Mike Sanderson, and Betsy Wertheim for fruitful conversations and insight. Funding: JOW acknowledges funding from the National Institutes of Health (AI135992 and AI136056). JP acknowledges funding from the National Institutes of Health (T15LM011271) and the Google Cloud COVID-19 Research Credits Program. MW was supported by the David and Lucile Packard Foundation as well as the University of Arizona College of Science. NM acknowledges funding from the National Science Foundation (2028040) and the Google Cloud COVID-19 Research Credits Program. Author contributions: Conceptualization: JOW; Methodology: JP, NM, MW, KS, JOW; Software: JP, NM, JOW; Validation: JP, JOW; Formal analysis: JP, JOW; Investigation: JP, JOW; Resources: JOW; Data Curation: JOW; Writing – original draft preparation: JP, JOW; Writing – review and editing: JP, MW, KS, JOW, NM; Visualization: JP, JOW; Supervision: JOW; Project administration: JOW; Funding acquisition: JOW, MW. Competing interests: JOW has received funding from Gilead Sciences, LLC (completed) and the CDC (ongoing) via grants and contracts to his institution unrelated to this research. Data and materials availability: All data used in this analysis are free to access via GISAID. BEAST XML input, FAVITES JSON, and results files are available at DataDryad.org (https://doi.org/10.5061/dryad.4f4qrfjbm) (44). This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) license, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. To view a copy of this license, visit https://creativecommons.org/licenses/by/4.0. This license does not apply to figures/photos/artwork or other content included in the article that is credited to a third party; obtain authorization from the rights holder before using such material.“
https://science.sciencemag.org/content/early/2021/03/17/science.abf8003